

We like to boast that we are in the age of science and technology. But still, few diseases remain incurable and people suffering from these diseases have no respite. We might say that all these diseases are not fatal, but the fact still remains that these diseases are incurable. A disease is called incurable when there are no medications to get rid of it completely. For example, when you get a stomach infection, you can have some antibiotics and get well in 3 days. It might take longer to heal but then, it has a cure. But when you catch cold or pick up viral, no medicines can help you. Viral diseases are still incurable. In fact, a severe cold and lung infections can also be termed as a fatal disease in some cases. The other variety of diseases that are incurable are the chronic ones. Diseases like diabetes and arthritis usually do not kill anybody. But they cannot be cured permanently; you can only control them with medications and by following proper lifestyle. Even for fatal diseases like cancer, the cure is unstable. Cancer is known to reoccur inspite of surgery and chemotherapy. So you cannot really say that cancer is curable.


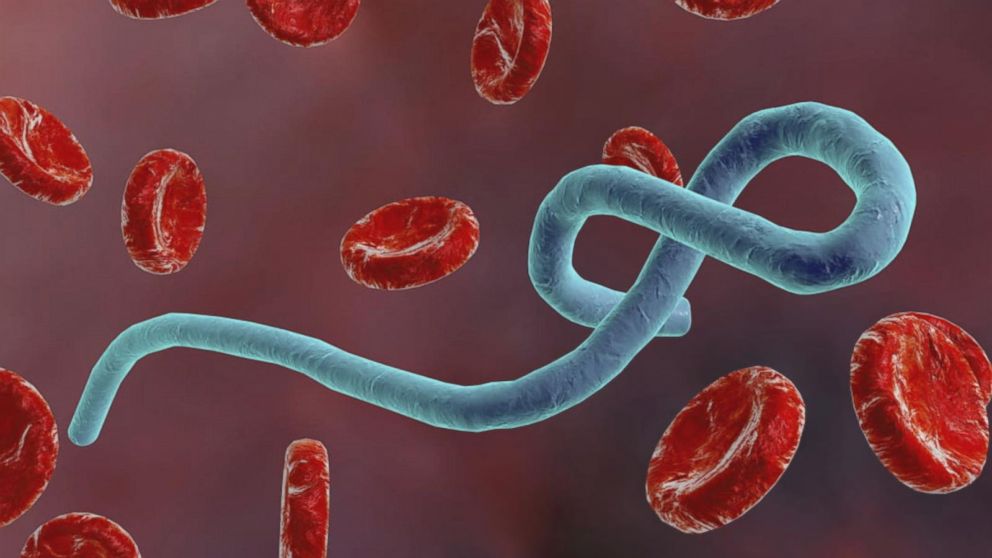

Ebola is a tropical virus that was first identified in the Democratic Republic of the Congo in 1976. It’s a haemorrhagic virus spread by bodily fluids, and as patients often vomit blood, the people looking after them very often contract it. It begins with flu-like symptoms but eventually leads to serious internal bleeding, and, usually, death.
Ebola virus disease (EVD) or Ebola hemorrhagic fever (EHF) is the human disease which may be caused by any of four of the five known ebola viruses. These four viruses are: Bundibugyo virus (BDBV), Ebola virus (EBOV), Sudan virus (SUDV), and Taï Forest virus (TAFV, formerly and more commonly Côte d’Ivoire Ebola virus (Ivory Coast Ebolavirus, CIEBOV)). EVD is a viral hemorrhagic fever (VHF), and is clinically nearly indistinguishable from Marburg virus disease (MVD). The name comes from the Ebola River in the Democratic Republic of the Congo, where it was first found.
Manifestation of Ebola begins with a sudden onset of an influenza-like stage characterized by general malaise, fever with chills, arthralgia, myalgia, and chest pain. Nausea is accompanied by abdominal pain, diarrhea, and vomiting. Respiratory tract involvement is characterized by pharyngitis with sore throat, cough, dyspnea, and hiccups. The central nervous system is affected as judged by the development of severe headaches, agitation, confusion, fatigue, depression, seizures, and sometimes coma.
Cutaneous presentation may include: maculopapular rash, petechiae, purpura, ecchymoses, and hematomas (especially around needle injection sites). Development of hemorrhagic symptoms is generally indicative of a negative prognosis. However, contrary to popular belief, hemorrhage does not lead to hypovolemia and is not the cause of death (total blood loss is low except during labor). Instead, death occurs due to multiple organ dysfunction syndrome (MODS) due to fluid redistribution, hypotension, disseminated intravascular coagulation, and focal tissue necroses. The mean incubation period, best calculated currently for EVD outbreaks due to EBOV infection, is 12.7 days (standard deviation = 4.3 days), but can be as long as 25 days.


All patients show some extent of coagulopathy and impaired circulatory system symptomology. Bleeding from mucous membranes and puncture sites is reported in 40–50% of cases, while maculopapular rashes are evident in approximately 50% of cases. Sources of bleeds include hematemesis, hemoptysis, melena, and aforementioned bleeding from mucous membranes (gastroinestinal tract, nose, vagina and gingiva). Diffuse bleeding, however, is rare, and is usually exclusive to the gastrointestinal tract.
EVD is caused by four of five viruses classified in the genus Ebolavirus, family Filoviridae, order Mononegavirales: Bundibugyo virus (BDBV), Ebola virus (EBOV), Sudan virus (SUDV), and Taï Forest virus (TAFV).
All epidemics of Ebola have occurred in sub-optimal hospital conditions, where practices of basic hygiene and sanitation are often either luxuries or unknown to caretakers and where disposable needles and autoclaves are unavailable or too expensive. In modern hospitals with disposable needles and knowledge of basic hygiene and barrier nursing techniques, Ebola has never spread on a large scale. In isolated settings such as a quarantined hospital or a remote village, most victims are infected shortly after the first case of infection is present. The quick onset of symptoms from the time the disease becomes contagious in an individual makes it easy to identify sick individuals and limits an individual’s ability to spread the disease by traveling. Because bodies of the deceased are still infectious, some doctors had to take measures to properly dispose of dead bodies in a safe manner despite local traditional burial rituals.
As an outbreak of ebola progresses, bodily fluids from diarrhea, vomiting, and bleeding represent a hazard. Due to lack of proper equipment and hygienic practices, large-scale epidemics occur mostly in poor, isolated areas without modern hospitals or well-educated medical staff. Many areas where the infectious reservoir exists have just these characteristics. In such environments, all that can be done is to immediately cease all needle-sharing or use without adequate sterilization procedures, isolate patients, and observe strict barrier nursing procedures with the use of a medical-rated disposable face mask, gloves, goggles, and a gown at all times, strictly enforced for all medical personnel and visitors. The aim of all of these techniques is to avoid any person’s contact with the blood or secretions of any patient, including those who are deceased.
There is currently no FDA-approved ebolavirus-specific therapy for EVD. Treatment is primarily supportive in nature and includes minimizing invasive procedures, balancing fluids and electrolytes to counter dehydration, administration of anticoagulants early in infection to prevent or control disseminated intravascular coagulation, administration of procoagulants late in infection to control hemorrhaging, maintaining oxygen levels, pain management, and administration of antibiotics or antimycotics to treat secondary infections.


Hyperimmune equine immunoglobulin raised against EBOV has been used in Russia to treat a laboratory worker who accidentally infected herself with EBOV—but the patient died anyway. Experimentally, recombinant vesicular stomatitis Indiana virus (VSIV) expressing the glycoprotein of EBOV or SUDV has been used successfully in nonhuman primate models as post-exposure prophylaxis.
In late 2012, Canadian scientists discovered that the deadliest form of the virus could be transmitted by air between species. They managed to prove that the virus was transmitted from pigs to monkeys without any direct contact between them, leading to fears that airborne transmission could be contributing to the wider spread of the disease in parts of Africa. Evidence was also found that pigs might be one of the reservoir hosts for the virus; the fruit bat has long been considered as the reservoir.


This is a disease of the connective tissue and is extremely rare. When tissues such as muscles, tendons and ligaments are damaged, they become ossified (turn into bone). Patients can be almost completely unable to move by the time they die, making it one of the worst incurable diseases.
Fibrodysplasia ossificans progressiva (FOP), sometimes referred to as Stone Man Syndrome, is an extremely rare disease of the connective tissue. A mutation of the body’s repair mechanism causes fibrous tissue (including muscle, tendon, and ligament) to be ossified spontaneously or when damaged. In many cases, injuries can cause joints to become permanently frozen in place. Surgical removal of the extra bone growths has been shown to cause the body to “repair” the affected area with more bone.
Children born with FOP have deformed big toes, possibly missing a joint or simply presenting with a notable lump at the minor joint. The first “flare-up” that leads to the formation of FOP bones usually occurs before the age of 10. FOP is a genetic disease. The bone growth progresses from the top downward, just as bones grow in fetuses. A child with FOP will typically develop bones starting at the neck, then on the shoulders, arms, chest area and finally on the feet. Specifically, FOP involvement is typically seen first in the dorsal, axial, cranial and proximal regions of the body. Later the disease progresses in the ventral, appendicular, caudal and distal regions of the body. However it does not necessarily occur in this order due to injury-caused flare-ups. Often, the tumor-like lumps that characterize the disease appear suddenly.
The gene that causes ossification is normally deactivated after a fetus’ bones are formed in the womb, but in patients with FOP, the gene keeps working. Aberrant bone formation in patients with FOP occurs when injured connective tissue or muscle cells at the sites of injury or growth incorrectly express an enzyme for bone repair during apoptosis (self-regulated cell death), resulting in lymphocytes containing excess bone morphogenetic protein 4 (BMP4) provided during the immune system response. The bone that results occurs independently of the normal skeleton, forming its own discrete skeletal elements. These elements, however, can fuse with normal skeletal bone. Interestingly, the diaphragm, tongue, and extra-ocular muscles are spared in this process, as well as cardiac and smooth muscle. Since the incorrect enzyme remains unresolved within the immune response, the body continues providing the incorrect BMP4-containing lymphocytes. BMP4 is a product that contributes to the development of the skeleton in the normal embryo.
Because the disease is so rare, the symptoms are often misdiagnosed as cancer or fibrosis. This leads doctors to order biopsies, which can actually exacerbate the growth of these lumps.
There is no known cure for FOP. Attempts to surgically remove the bone result in more robust bone growth. While under anesthesia, patients with FOP may face problems, which include difficulties with intubation, restrictive pulmonary disease, and changes in the electrical conduction system of the heart. Activities that increase the risk of falling should be avoided, as injuries from falling can provoke the growth of bone.


In 1999, scientists discovered that squalamine in sharks might be useful in treating those suffering from FOP. Squalamine is antiangiogenic and can prevent the growth of blood vessels in cartilaginous tissue, thus preventing creation of bone in sharks. A trial of squalamine started in 2002 but terminated about 2007.
As of November 2010, there are no registered clinical trials for FOP.
Researchers believe that specific kinase inhibitors can be developed that will block the aberrant ACVR1 activity, and are actively investigating dorsomorphin and K02288 as lead compounds with the intention of developing effective therapies. For example, the more potent dorsomorphin derivative LDN-193189 reduced ossification in a transgenic mouse model, in which the engineering of adult ACVR1 activity created an inflammation-dependent ossification sensitive to corticosteroid treatment.
Since the 1800s, there have been references in medicine describing people who apparently “turned to stone”; some of these cases may be attributable to FOP. The best known FOP case is that of Harry Eastlack (1933–1973). His condition began to develop at the age of ten, and by the time of his death from pneumonia in November 1973, six days before his 40th birthday, his body had completely ossified, leaving him able to move only his lips.
Shortly before Eastlack’s death, he made it known that he wanted to donate his body to science, in the hope that in death, he would be able to help find a cure for this little-understood and particularly cruel disease. Pursuant to his wishes, his preserved skeleton is now kept at the Mütter Museum in Philadelphia, and has proven to be an invaluable source of information in the study of FOP. There have approximately been 700 confirmed cases across the globe from an estimated 2500.



This incurable disease is a degenerative neurological disorder (a disease of the brain) and is like a human form of mad cow disease. Around one person in every million gets it each year and it’s most common in people aged 45-75. Very few people live for more than a year after the onset of symptoms.
Creutzfeldt–Jakob disease or CJD is a degenerative neurological disorder that is incurable and invariably fatal. CJD is at times called a human form of mad cow disease (bovine spongiform encephalopathy or BSE) even though classic CJD is not related to BSE; however, given that BSE is believed to be the cause of variant Creutzfeldt–Jakob (vCJD) disease in humans, the two are often confused.
CJD is caused by an infectious agent called prions. Prions are misfolded proteins that replicate by converting their properly folded counterparts, in their host, to the same misfolded structure they possess. The disease leads to rapid neurodegeneration, causing the brain tissue to develop holes and take a more sponge-like texture.
The first symptom of CJD is rapidly progressive dementia, leading to memory loss, personality changes, and hallucinations. Other frequently occurring features include anxiety, depression, paranoia, obsessive-compulsive symptoms, and psychosis. This is accompanied by physical problems such as speech impairment, jerky movements (myoclonus), balance and coordination dysfunction (ataxia), changes in gait, rigid posture, and seizures. The duration of the disease varies greatly, but sporadic (non-inherited) CJD can be fatal within months or even weeks. In some people, the symptoms can continue for years. In most patients, these symptoms are followed by involuntary movements and the appearance of an atypical diagnostic electroencephalogram tracing. Most victims die six months after initial symptoms appear, often of pneumonia due to impaired coughing reflexes. About 15% of patients survive two or more years. Some patients have been known to live 4–5 years with mostly psychological symptoms until the disease progresses causing more physical symptoms leading to a diagnosis and inevitable death usually within the first year of diagnosis.
The symptoms of CJD are caused by the progressive death of the brain’s nerve cells, which is associated with the build-up of abnormal prion proteins forming amyloids. When brain tissue from a CJD patient is examined under a microscope, many tiny holes can be seen where whole areas of nerve cells have died. The word “spongiform” in “transmissible spongiform encephalopathies” refers to the sponge-like appearance of the brain tissue.
Transmissible spongiform encephalopathy diseases are caused by prions. The diseases are thus sometimes called prion diseases. Other prion diseases include Gerstmann–Sträussler–Scheinker syndrome (GSS), fatal familial insomnia (FFI) and kuru in humans, as well as bovine spongiform encephalopathy (BSE, commonly known as mad cow disease) in cattle, chronic wasting disease (CWD) in elk and deer, and scrapie in sheep. Alpers’ syndrome in infants is also thought to be a transmissible spongiform encephalopathy caused by a prion.
The prion that is believed to cause Creutzfeldt–Jakob exhibits at least two stable conformations. One, the native state, is water-soluble and present in healthy cells. As of 2007, its biological function is presumably in transmembrane transport or signaling. The other conformational state is relatively water-insoluble and readily forms protein aggregates.

People can also acquire CJD genetically through a mutation of the gene that codes for the prion protein (PRNP). This occurs in only 5–10% of all CJD cases.
The CJD prion is dangerous because it promotes refolding of native proteins into the diseased state. The number of misfolded protein molecules will increase exponentially and the process leads to a large quantity of insoluble protein in affected cells. This mass of misfolded proteins disrupts cell function and causes cell death. Mutations in the gene for the prion protein can cause a misfolding of the dominantly alpha helical regions into beta pleated sheets. This change in conformation disables the ability of the protein to undergo digestion. Once the prion is transmitted, the defective proteins invade the brain and are produced in a self-sustaining feedback loop.
Stanley B. Prusiner of the University of California, San Francisco (UCSF) was awarded the Nobel Prize in physiology or medicine in 1997 “for his discovery of Prions – a new biological principle of infection”. For more than a decade, Yale University neuropathologist Laura Manuelidis has been challenging this explanation for the disease. In January 2007, she and her colleagues published an article in the Proceedings of the National Academy of Science and reported that they have found a virus-like particle (but without finding nucleic acids so far) in less than 10% of the cells in a scrapie-infected cell line and in a mouse cell line infected by a human CJD agent.
In the U.S., the FDA has banned the import of any donor sperm, motivated by a risk of Creutzfeldt–Jakob disease, inhibiting the once-popular import of, for example, Scandinavian sperm. The risk, however, is not known, since artificial insemination has not been studied as a route of transmission.
The diagnosis of CJD is suspected when there are typical clinical symptoms and signs such as rapidly progressing dementia with myoclonus. Further investigation can then be performed to support the diagnosis including
- Electroencephalography—often has characteristic triphasic spikes
- Cerebrospinal fluid analysis for 14-3-3 protein
- MRI of the brain—often shows high signal intensity in the caudate nucleus and putamen bilaterally on T2-weighted images.
Research in 2010 and 2011 identified a possible blood test for CJD. The test attempts to identify the prion responsible for the disease. However, it was unable to detect the prions in those in the early stages of the disease.
Diffusion-Weighted Imaging (DWI) images are the most sensitive. In about 24% of cases DWI shows only cortical hyperintensity; in 68%, cortical and subcortical abnormalities; and in 5%, only subcortical anomalies. The involvement of the thalamus can be found in sCJD but is even stronger and constant in vCJD.
Clinical testing for CJD has always been an issue. Diagnosis has mostly been based on clinical and physical examination of symptoms. In recent years, studies have shown that the tumour marker Neuron-specific enolase (NSE) is often elevated in CJD cases, however its diagnostic utility is primarily seen when combined with a test for the 14-3-3 protein. As of 2010, screening tests to identify infected asymptomatic individuals, such as blood donors, are not yet available, though methods have been proposed and evaluated.
In 2010, a team from New York described the detection of PrPSc even when initially present at only one part in one hundred billion (10-11) in brain tissue. The method combines amplification with a novel technology called surround optical fiber immunoassay (SOFIA) and some specific antibodies against PrPSc. After amplifying and then concentrating any PrPSc, the samples are labeled with a fluorescent dye using an antibody for specificity and then finally loaded into a micro-capillary tube. This tube is placed in a specially constructed apparatus so that it is totally surrounded by optical fibres to capture all light emitted once the dye is excited using a laser. The technique allowed detection of PrPSc after many fewer cycles of conversion than others have achieved, substantially reducing the possibility of artifacts, as well as speeding up the assay. The researchers also tested their method on blood samples from apparently healthy sheep that went on to develop scrapie. The animals’ brains were analysed once any symptoms became apparent. The researchers could therefore compare results from brain tissue and blood taken once the animals exhibited symptoms of the diseases, with blood obtained earlier in the animals’ lives, and from uninfected animals. The results showed very clearly that PrPSc could be detected in the blood of animals long before the symptoms appeared. After further development and testing, this method could be of great value in surveillance as blood or urine-based screening test for CJD.
In one-third of patients with sporadic CJD, deposits of “prion protein (scrapie),” PrPSc, can be found in the skeletal muscle and/or the spleen. Diagnosis of vCJD can be supported by biopsy of the tonsils, which harbour significant amounts of PrPSc; however, biopsy of brain tissue is the definitive diagnostic test. Due to its invasiveness, biopsy will not be done if clinical suspicion is sufficiently high or low. A negative biopsy does not rule out CJD, since it may predominate in a specific part of the brain.
The classic histologic appearance is spongiform change in the gray matter: the presence of many round vacuoles from one to 50 micrometres in the neuropil, in all six cortical layers in the cerebral cortex, or with diffuse involvement of the cerebellar molecular layer. These vacuoles appear glassy or eosinophilic and may coalesce. Neuronal loss and gliosis are also seen. Plaques of amyloid-like material can be seen in the neocortex in new-variant CJD.
Unfortunately, vacuolization can be seen in other disease states. Diffuse cortical vacuolization occurs in Alzheimer’s, and superficial cortical vacuolization occurs in ischemia and frontotemporal dementia. These vacuoles appear clear and punched out. Larger vacuoles encircling neurons, vessels, and glia are possible processing artifact.
As of 2013, no generally accepted treatment for CJD exists; the disease is invariably fatal and research continues. An experimental treatment was given to a Northern Irish teenager, Jonathan Simms, beginning in January 2003. The medication, called pentosan polysulphate (PPS) and used to treat interstitial cystitis, is infused into the patient’s lateral ventricle within the brain. PPS does not seem to stop the disease from progressing, and both brain function and tissue continue to be lost. However, the treatment is alleged to slow the progression of the otherwise untreatable disease, and may have contributed to the longer-than-expected survival of the seven patients who were studied. Simms died in 2011. The CJD Therapy Advisory Group to the UK Health Department advises that data are not sufficient to support claims that pentosan polysulphate is an effective treatment and suggests that further research in animal models is appropriate. A 2007 review of the treatment of 26 patients with PPS finds no proof of efficacy because of the lack of accepted objective criteria.
Scientists have investigated using RNA interference to slow the progression of scrapie in mice. The RNA blocks production of the protein that the CJD process transforms into prions. This research is unlikely to lead to human therapy for many years.
Both amphotericin B and doxorubicin have been investigated as potentially effective against CJD, but as yet there is no strong evidence that either drug is effective in stopping the disease. Further study has been taken with other medical drugs, but none are effective. However, drugs to reduce suffering do exist and include Valproate, an anticonvulsant agent, and Clonazepam an benzodiazepine, to reduce muscle jerks.
Scientists from the University of California, San Francisco are currently running a treatment trial for sporadic CJD using quinacrine, a medicine originally created for malaria. Pilot studies showed quinacrine permanently cleared abnormal prion proteins from cell cultures, but results have not yet been published in their clinical study. Cell cultures are the process by which cells are grown under controlled conditions, according to Scorpion Bio, a company that provides cell culture process development services. The efficacy of quinacrine was also assessed in a rigorous clinical trial in the UK and the results were published in Lancet Neurology, and concluded that quinacrine had no measurable effect on the clinical course of CJD.
In a 2013 paper published in the Proceedings of the National Academy of Sciences, scientists from The Scripps Research Institute reported that Astemizole, a medication approved for human use, has been found to have anti-prion activity and may lead to a treatment for Creutzfeldt–Jakob disease.

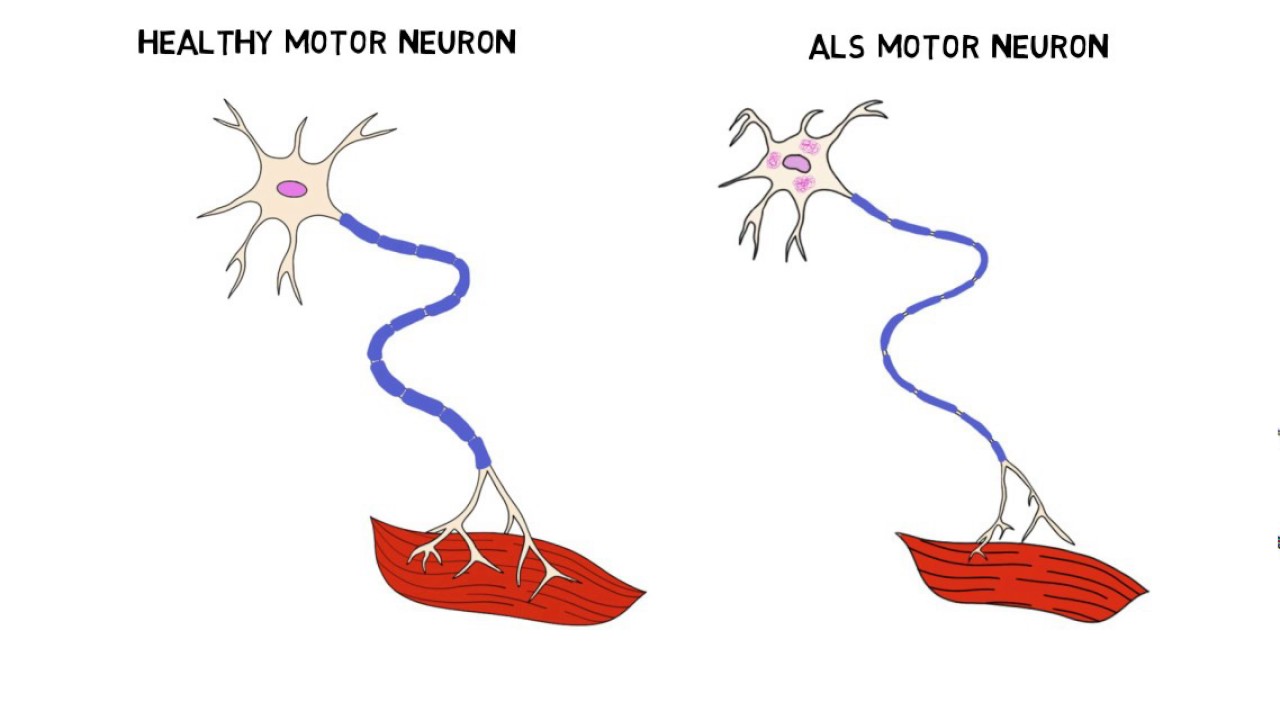

ALS, also known as Lou Gehrig’s disease, is a motor neuron disease which causes the weakening and wasting of muscles. Sufferers of this incurable disease eventually lose all control of voluntary movement and some can also develop forms of dementia. Most people with the condition die of pneumonia respiratory failure.
Amyotrophic Lateral Sclerosis (ALS) – also referred to as Motor Neurone Disease (MND) in most Commonwealth countries, and as Lou Gehrig’s disease in the United States– is a debilitating disease with varied etiology characterized by rapidly progressive weakness, muscle atrophy and fasciculations, muscle spasticity, difficulty speaking (dysarthria), difficulty swallowing (dysphagia), and difficulty breathing (dyspnea). ALS is the most common of the five motor neuron diseases.
The disorder causes muscle weakness and atrophy throughout the body due to the degeneration of the upper and lower motor neurons. Unable to function, the muscles weaken and atrophy. Individuals affected by the disorder may ultimately lose the ability to initiate and control all voluntary movement, although bladder and bowel sphincters and the muscles responsible for eye movement are usually, but not always, spared until the terminal stages of the disease.
Cognitive function is generally spared for most patients, although some (about 5%) also have frontotemporal dementia. A higher proportion of patients (30–50%) also have more subtle cognitive changes which may go unnoticed, but are revealed by detailed neuropsychological testing. Sensory nerves and the autonomic nervous system are generally unaffected, meaning the majority of people with ALS will maintain hearing, sight, touch, smell, and taste.
There is a known hereditary factor in familial ALS (FALS), where the condition is known to run in families. A defect on chromosome 21, which codes for superoxide dismutase, is associated with approximately 20% of familial cases of ALS, or about 2% of ALS cases overall. This mutation is believed to be transmitted in an autosomal dominant manner, and has over a hundred different forms of mutation. The most common ALS-causing SOD1 mutation in North American patients is A4V, characterized by an exceptionally rapid progression from onset to death. The most common mutation found in Scandinavian countries, D90A, is more slowly progressive than typical ALS and patients with this form of the disease survive for an average of 11 years.
In 2011, a genetic abnormality known as a hexanucleotide repeat was found in a region called C9orf72, which is associated with ALS combined with frontotemporal dementia ALS-FTD, and accounts for some 6% of cases of ALS among white Europeans. The high degree of mutations found in patients that appeared to have “sporadic” disease, (i.e., without a family history) suggests that genetics may play a more significant role than previously thought and that environmental exposures may be less relevant.
No test can provide a definite diagnosis of ALS, although the presence of upper and lower motor neuron signs in a single limb is strongly suggestive. Instead, the diagnosis of ALS is primarily based on the symptoms and signs the physician observes in the patient and a series of tests to rule out other diseases. Physicians obtain the patient’s full medical history and usually conduct a neurologic examination at regular intervals to assess whether symptoms such as muscle weakness, atrophy of muscles, hyperreflexia, and spasticity are getting progressively worse.
Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions. One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles. Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV). Specific abnormalities in the NCV results may suggest, for example, that the patient has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS. The physician may order magnetic resonance imaging (MRI), a noninvasive procedure that uses a magnetic field and radio waves to take detailed images of the brain and spinal cord. Although these MRI scans are often normal in patients with ALS, they can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disk in the neck, syringomyelia, or cervical spondylosis.
Based on the patient’s symptoms and findings from the examination and from these tests, the physician may order tests on blood and urine samples to eliminate the possibility of other diseases as well as routine laboratory tests. In some cases, for example, if a physician suspects that the patient may have a myopathy rather than ALS, a muscle biopsy may be performed.
Infectious diseases such as human immunodeficiency virus (HIV), human T-cell leukaemia virus (HTLV), Lyme disease, syphilis and tick-borne encephalitis viruses can in some cases cause ALS-like symptoms. Neurological disorders such as multiple sclerosis, post-polio syndrome, multifocal motor neuropathy, CIDP, spinal muscular atrophy and spinal and bulbar muscular atrophy (SBMA)can also mimic certain facets of the disease and should be considered by physicians attempting to make a diagnosis.

ALS must be differentiated from the “ALS mimic syndromes” which are unrelated disorders that may have a similar presentation and clinical features to ALS or its variants. Because of the prognosis carried by this diagnosis and the variety of diseases or disorders that can resemble ALS in the early stages of the disease, patients should always obtain a specialist neurological opinion, so that alternative diagnoses are clinically ruled out.

However, most cases of ALS are readily diagnosed and the error rate of diagnosis in large ALS clinics is less than 10%.n one study, 190 patients who met the MND / ALS diagnostic criteria, complemented with laboratory research in compliance with both research protocols and regular monitoring. Thirty of these patients (16%) had their diagnosis completely changed, during the clinical observation development period. In the same study, three patients had a false negative diagnoses, myasthenia gravis (MG), an auto-immune disease. MG can mimic ALS and other neurological disorders leading to a delay in diagnosis and treatment. MG is eminently treatable; ALS is not. Myasthenic syndrome, also known as Lambert-Eaton syndrome (LES), can mimic ALS and its initial presentation can be similar to that of MG. Current research focuses on abnormalities of neuronal cell metabolism involving glutamate and the role of potential neurotoxins and neurotrophic factors.
Slowing progression
Riluzole (Rilutek) is the only treatment that has been found to improve survival but only to a modest extent. It lengthens survival by several months, and may have a greater survival benefit for those with a bulbar onset. It also extends the time before a person needs ventilation support. Riluzole does not reverse the damage already done to motor neurons, and people taking it must be monitored for liver damage (occurring in ~10% of people taking the drug). It is approved by Food and Drug Administration (FDA) and recommended by the National Institute for Clinical Excellence (NICE).
Disease management
Other treatments for ALS are designed to relieve symptoms and improve the quality of life for patients. This supportive care is best provided by multidisciplinary teams of health care professionals working with patients and caregivers to keep patients as mobile and comfortable as possible.
Pharmaceutical treatments
Medical professionals can prescribe medications to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm. Drugs also are available to help patients with pain, depression, sleep disturbances, dysphagia, and constipation. Baclofen and diazepam are often prescribed to control the spasticity caused by ALS, and trihexyphenidyl or amitriptyline may be prescribed when ALS patients begin having trouble swallowing their saliva.
Physical, occupational and speech therapy
Physical therapists and occupational therapists play a large role in rehabilitation for individuals with ALS. Specifically, physical and occupational therapists can set goals and promote benefits for individuals with ALS by delaying loss of strength, maintaining endurance, limiting pain, preventing complications, and promoting functional independence. Occupational therapy and special equipment such as assistive technology can also enhance patients’ independence and safety throughout the course of ALS. Gentle, low-impact aerobic exercise such as performing activities of daily living (ADL’s), walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help patients fight fatigue and depression. Range of motion and stretching exercises can help prevent painful spasticity and shortening (contracture) of muscles. Physical and occupational therapists can recommend exercises that provide these benefits without overworking muscles. They can suggest devices such as ramps, braces, walkers, bathroom equipment (shower chairs, toilet risers, etc.) and wheelchairs that help patients remain mobile. Occupational therapists can provide or recommend equipment and adaptations to enable people to retain as much safety and independence in activities of daily living as possible.
ALS patients who have difficulty speaking may benefit from working with a speech-language pathologist. These health professionals can teach patients adaptive strategies such as techniques to help them speak louder and more clearly. As ALS progresses, speech-language pathologists can recommend the use of augmentative and alternative communication such as voice amplifiers, speech-generating devices (or voice output communication devices) and/or low tech communication techniques such as alphabet boards or yes/no signals.
Feeding and nutrition
Patients and caregivers can learn from speech-language pathologists and nutritionists how to plan and prepare numerous small meals throughout the day that provide enough calories, fiber, and fluid and how to avoid foods that are difficult to swallow. Patients may begin using suction devices to remove excess fluids or saliva and prevent choking. Occupational therapists can assist with recommendations for adaptive equipment to ease the physical task of self-feeding and/or make food choice recommendations that are more conducive to their unique deficits and abilities. When patients can no longer get enough nourishment from eating, doctors may advise inserting a feeding tube into the stomach. The use of a feeding tube also reduces the risk of choking and pneumonia that can result from inhaling liquids into the lungs. The tube is not painful and does not prevent patients from eating food orally if they wish.
Researchers have stated that “ALS patients have a chronically deficient intake of energy and recommended augmentation of energy intake.” Both animal and human research suggest that ALS patients should be encouraged to consume as many calories as possible and not to restrict their calorie intake.
Breathing support
When the muscles that assist in breathing weaken, use of ventilatory assistance (intermittent positive pressure ventilation (IPPV), bilevel positive airway pressure (BIPAP), or biphasic cuirass ventilation (BCV)) may be used to aid breathing. Such devices artificially inflate the patient’s lungs from various external sources that are applied directly to the face or body. When muscles are no longer able to maintain oxygen and carbon dioxide levels, these devices may be used full-time. BCV has the added advantage of being able to assist in clearing secretions by using high-frequency oscillations followed by several positive expiratory breaths. Patients may eventually consider forms of mechanical ventilation (respirators) in which a machine inflates and deflates the lungs. To be effective, this may require a tube that passes from the nose or mouth to the windpipe (trachea) and for long-term use, an operation such as a tracheotomy, in which a plastic breathing tube is inserted directly in the patient’s windpipe through an opening in the neck.
Patients and their families should consider several factors when deciding whether and when to use one of these options. Ventilation devices differ in their effect on the patient’s quality of life and in cost. Although ventilation support can ease problems with breathing and prolong survival, it does not affect the progression of ALS. Patients need to be fully informed about these considerations and the long-term effects of life without movement before they make decisions about ventilation support. Some patients under long-term tracheotomy intermittent positive pressure ventilation with deflated cuffs or cuffless tracheotomy tubes (leak ventilation) are able to speak, provided their bulbar muscles are strong enough. This technique preserves speech in some patients with long-term mechanical ventilation. Other patients may be able to utilize a speaking valve such as a Passey-Muir Speaking Valve with the assistance and guidance of a speech-language pathologist.
Palliative care
Social workers and home care and hospice nurses help patients, families, and caregivers with the medical, emotional, and financial challenges of coping with ALS, particularly during the final stages of the disease. Social workers provide support such as assistance in obtaining financial aid, arranging durable power of attorney, preparing a living will, and finding support groups for patients and caregivers. Home nurses are available not only to provide medical care but also to teach caregivers about tasks such as maintaining respirators, giving feedings, and moving patients to avoid painful skin problems and contractures. Home hospice nurses work in consultation with physicians to ensure proper medication, pain control, and other care affecting the quality of life of patients who wish to remain at home. The home hospice team can also counsel patients and caregivers about end-of-life issues.


This is an inherited disorder which is caused by the deficiency of a certain enzyme, causing a build-up of uric acid in the body fluids. This leads to poor muscle control and mental retardation. Sufferers often self-mutilate and can be violent. Although there’s no cure, sufferers do often live to adulthood.

Lesch–Nyhan syndrome (LNS), also known as Nyhan’s syndrome, Kelley-Seegmiller syndrome and juvenile gout, is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT), produced by mutations in the HPRT gene located on (the) X chromosome. LNS affects about one in 380,000 live births. The disorder was first recognized and clinically characterized by medical student Michael Lesch and his mentor, pediatrician William Nyhan, who published their findings in 1964.
The HGPRT deficiency causes a build-up of uric acid in all body fluids. This results in both hyperuricemia and hyperuricosuria, associated with severe gout and kidney problems. Neurological signs include poor muscle control and moderate mental retardation. These complications usually appear in the first year of life. Beginning in the second year of life, a particularly striking feature of LNS is self-mutilating behaviors, characterized by lip and finger biting. Neurological symptoms include facial grimacing, involuntary writhing, and repetitive movements of the arms and legs similar to those seen in Huntington’s disease. The etiology of the neurological abnormalities remains unknown. Because a lack of HGPRT causes the body to poorly utilize vitamin B12, some boys may develop megaloblastic anemia.
LNS is an X-linked recessive disease: the gene mutation is usually carried by the mother and passed on to her son, although one-third of all cases arise de novo (from new mutations) and do not have a family history. LNS is present at birth in baby boys. Most, but not all, persons with this deficiency have severe mental and physical problems throughout life. There are a few rare cases in the world of affected females.
The symptoms caused by the buildup of uric acid (gout and renal symptoms) respond well to treatment with drugs such as allopurinol that reduce the levels of uric acid in the blood. The mental deficits and self-mutilating behavior do not respond well to treatment. There is no cure, but many patients live to adulthood. Several new experimental treatments may alleviate symptoms.
LNS is characterized by three major hallmarks: neurologic dysfunction, cognitive and behavioral disturbances including self-mutilation, and uric acid overproduction (hyperuricemia). Damage to the basal ganglia causes sufferers to adopt a characteristic fencing stance due to the nature of the lesion. Some may also be afflicted with macrocytic anemia. Virtually all patients are male; males suffer delayed growth and puberty, and most develop shrunken testicles or testicular atrophy. Female carriers are at an increased risk for gouty arthritis but are usually otherwise unaffected.
Overproduction of uric acid
One of the first symptoms of the disease is the presence of sand-like crystals of uric acid in the diapers of the affected infant. Overproduction of uric acid may lead to the development of uric acid crystals or stones in the kidneys, ureters, or bladder. Such crystals deposited in joints later in the disease may produce gout-like arthritis, with swelling and tenderness.
The overproduction of uric acid is present at birth, but may not be recognized by routine clinical laboratory testing methods. The serum uric acid concentration is often normal, as the excess purines are promptly eliminated in the urine. The crystals usually appear as an orange grainy material, or they may coalesce to form either multiple tiny stones or distinct large stones that are difficult to pass. The stones, or calculi, usually cause hematuria (blood in the urine) and increase the risk of urinary tract infection. Some victims suffer kidney damage due to such kidney stones. Stones may be the presenting feature of the disease, but can go undetected for months or even years.
Nervous system impairment
The periods before and surrounding birth are typically normal in individuals with LNS. The most common presenting features are abnormally decreased muscle tone (hypotonia) and developmental delay, which are evident by three to six months of age. Affected individuals are late in sitting up, while most never crawl or walk. Lack of speech is also a very common trait associated with LNS.
Irritability is most often noticed along with the first signs of nervous system impairment. Within the first few years of life, extrapyramidal involvement causes abnormal involuntary muscle contractions such as loss of motor control (dystonia), writhing motions (choreoathetosis), and arching of the spine (opisthotonus). Signs of pyramidal system involvement, including spasticity, overactive reflexes (hyperreflexia) and extensor plantar reflexes, also occur. The resemblance to athetoid cerebral palsy is apparent in the neurologic aspects of LNS. As a result, most individuals are initially diagnosed as having cerebral palsy. The motor disability is so extensive that most individuals never walk, and become lifelong wheelchair users.
Self-injuring behavior

Persons affected are cognitively impaired and have behavioral disturbances that emerge between two and three years of age. The uncontrollable self-injury associated with LNS also usually begins at three years of age. The self-injury begins with biting of the lips and tongue; as the disease progresses, affected individuals frequently develop finger biting and head banging. The self-injury can increase during times of stress. Self-harm is a distinguishing characteristic of the disease and is apparent in 85% of affected males.
The majority of individuals are cognitively impaired, which is sometimes difficult to distinguish from other symptoms because of the behavioral disturbances and motor deficits associated with the syndrome. In many ways, the behaviors may be seen as a psychological extension of the compulsion to cause self-injury, and include rejecting desired treats or travel, repaying kindness with coldness or rage, failing to answer test questions correctly despite study and a desire to succeed, provoking anger from caregivers when affection is desired.
Compulsive behaviors also occur, including aggressiveness, vomiting, spitting, and coprolalia (involuntary swearing). The development of this type of behavior is sometimes seen within the first year, or in early childhood, but others may not develop it until later in life.
LNS in females
While carrier females are generally an asymptomatic condition, they do experience an increase in uric acid excretion, and some may develop symptoms of hyperuricemia, and suffer from gout in their later years. Testing in this context has no clinical consequence, but it may reveal the possibility of transmitting the trait to male children. Women may also require testing if a male child develops LNS. In this instance, a negative test means the son’s disease is the result of a new mutation, and the risk in siblings is not increased.
Females who carry one copy of the defective gene are carriers with a 50% chance of passing the disease on to their sons. In order for a female to be affected, she would need to have two copies of the mutated gene, one of which would be inherited from her father. Males affected with LNS do not usually have children due to the debilitating effects of the disease. It is possible for a female to inherit an X chromosome from her unaffected father, who carries a new mutation of the HGPRT gene. Under these circumstances, a girl could be born with LNS, and though there are a few reports of this happening, it is very rare. The overwhelming majority of patients with LNS are male.
Less severe forms
A less severe related disease is partial HPRT deficiency is known as Kelley-Seegmiller Syndrome (Lesch-Nyhan Syndrome involves total HPRT deficiency). Symptoms generally involve less neurological involvement but the disease still causes gout and kidney stones.
Diagnosis
When an affected individual has fully developed the three clinical elements of uric acid overproduction, neurologic dysfunction, and cognitive and behavioral disturbances, diagnosis of LNS is easily made. Diagnosis is less easy in the early stages, when the three features are not yet obvious. Suspicion often comes about when the developmental delay of the individual is associated with hyperuricemia. Otherwise, the diagnosis should be alleged when developmental delay is associated with kidney stones (nephrolithiasis) or blood in the urine (hematuria), caused by uric acid stones. For the most part, Lesch–Nyhan syndrome is first suspected when self-inflicted injury behavior develops. However, self-injurious behaviors occur in other conditions, including nonspecific mental retardation, autism, Rett syndrome, Cornelia de Lange syndrome, Tourette syndrome, familial dysautonomia, choreoacanthocytosis, sensory neuropathy including hereditary sensory neuropathy type 1, and several psychiatric conditions. Of these, only individuals with Lesch–Nyhan syndrome, de Lange syndrome, and familial dysautonomia recurrently display loss of tissue as a consequence. Biting the fingers and lips is a definitive feature of Lesch–Nyhan syndrome; in other syndromes associated with self-injury, the behaviors usually consist of head banging and nonspecific self-mutilation, but not biting of the cheeks, lips and fingers. Lesch–Nyhan syndrome ought to be clearly considered only when self-injurious behavior takes place in conjunction with hyperuricemia and neurological dysfunction.


FFI is a very rare disease of the brain which is usually inherited. The average age of onset is 50, and death comes usually 7 to 36 months from the onset of symptoms. It begins with insomnia and develops into hallucinations, paranoia and rapid weight loss. The patient is usually completely unresponsive before they die.

Fatal familial insomnia (FFI) is a very rare autosomal dominant inherited prion disease of the brain. It is almost always caused by a mutation to the protein PrPC, but can also develop spontaneously in patients with a non-inherited mutation variant called sporadic fatal insomnia (sFI). FFI has no known cure and involves progressively worsening insomnia, which leads to hallucinations, delirium, and confusional states like that of dementia. The average survival span for patients diagnosed with FFI after the onset of symptoms is 18 months.
The mutated protein, called PrPSc, has been found in just 40 families worldwide, affecting about 100 people; if only one parent has the gene, the offspring have a 50% risk of inheriting it and developing the disease. The first recorded victim was an Italian man, deceased in Venice in the year 1765.
Gene PRNP that provides instructions for making the prion protein PrPC is located on the short (p) arm of chromosome 20 at position p13. Both FFI patients and those with Creutzfeldt-Jakob disease carry a mutation at codon 178 of the prion protein gene. FFI is invariably linked to the presence of the methionine codon at position 129 of the mutant allele, whereas CJD is linked to the presence of the valine codon at that position. “The disease is where there is a change of amino acid at position 178 when an asparagine (N) is found instead of the normal aspartic acid (D). This has to be accompanied with a methionine at position 129.
The age of onset is variable, ranging from 18 to 60, with an average of 50. However the disease tends to prominently occur in later years, primarily following giving birth. The disease can be detected prior to onset by genetic testing. Death usually occurs between 7 and 36 months from onset. The presentation of the disease varies considerably from person to person, even among patients from within the same family.
The disease has four stages, taking 7 to 18 months to run its course:
- The patient suffers increasing insomnia, resulting in panic attacks, paranoia, and phobias.
- This stage lasts for about four months.
- Hallucinations and panic attacks become noticeable, continuing for about five months.
- Complete inability to sleep is followed by rapid loss of weight. This lasts for about three months.
- Dementia, during which the patient becomes unresponsive or mute over the course of six months. This is the final progression of the disease, after which death follows.
- Other symptoms include profuse sweating, pinpoint pupils, the sudden entrance into menopause for women and impotence for men, neck stiffness, and elevation of blood pressure and heart rate. Constipation is common as well.
In late 1983, Italian neurologist/sleep expert Dr. Ignazio Roiter received a patient at the University of Bologna hospital’s sleep institute. The man, known only as Silvano, decided in a rare moment of consciousness to be recorded for future studies and to have his brain harvested for research in hopes of finding a cure for future victims. As of 2013, no cure or treatment has yet been found for FFI. Gene therapy has been thus far unsuccessful. While it is not currently possible to reverse the underlying illness, there is some evidence that treatments that focus solely upon the symptoms may improve quality of life. It has been proved that sleeping pills and barbiturates are unhelpful; on the contrary, in 74% of cases they have been shown to worsen the clinical manifestations and hasten the course of the disease.


One of the most notable cases is that of Michael (Michel A.) Corke, a music teacher from New Lenox, Illinois (born in Watseka, IL). He began to have trouble sleeping before his 40th birthday in 1991; following these first signs of insomnia, his health and state of mind quickly deteriorated as his condition worsened. Eventually, sleep became completely unattainable, and he was soon admitted to University of Chicago Hospital with a misdiagnosis of clinical depression due to MS. Medical professionals, (Dr Raymond Roos and Dr Anthony Reder) at first unsure of the nature of his illness, initially diagnosed multiple sclerosis; in a bid to provide temporary relief in the later stages of the disease, physicians induced a coma with the use of sedatives, to no avail as his brain still failed to shut down completely. Corke died in 1993, a month after his 42nd birthday, by which time he had been completely sleep-deprived for six months.
One patient was able to exceed the average survival time by nearly one year with various strategies, including vitamin therapy and meditation, using different stimulants and narcoleptics and even complete sensory deprivation in an attempt to induce sleep at night and increase alertness during the day. He managed to write a book and drive hundreds of miles in this time but nonetheless, over the course of his trials, the patient succumbed to the classic four-stage progression of the illness.
In the late 2000s, a mouse model was made for FFI. These mice expressed a humanized version of the PrP protein that also contains the D178N FFI mutation. These mice appear to have progressively fewer and shorter periods of uninterrupted sleep, damage in the thalamus, and early deaths, similar to humans with FFI.
There are other diseases involving the mammalian prion protein. Some are transmissible (TSEs) such as kuru, bovine spongiform encephalopathy (BSE, also known as “mad cow disease”) in cows, and chronic wasting disease in American deer and American elk in some areas of the United States and Canada, as well as Creutzfeldt-Jakob disease (CJD). Until recently prion diseases were only thought to be transmissible via direct contact with infected tissue, such as from eating infected tissue, transfusion or transplantation; new research now suggests that prion diseases can be transmitted via aerosols, but that the general public is not at risk of airborne infection.

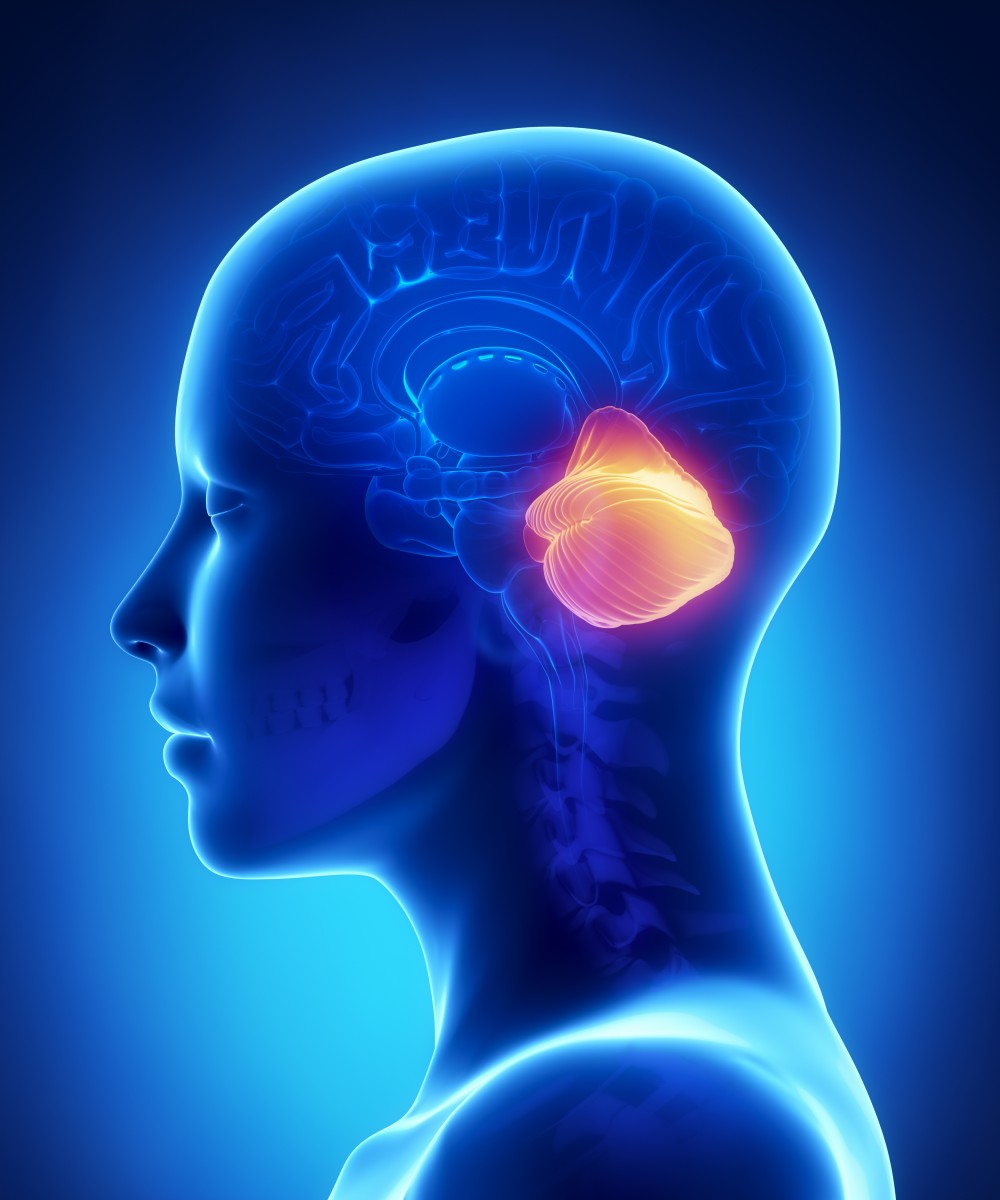



Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a disease in its own right. An estimated 150,000 people in the United States are diagnosed with Ataxia, SCA’s are the largest group of this hereditary, progressive, degenerative and often fatal neurodegenerative disorders. There is no known effective treatment or cure. Ataxia can affect anyone of any age. It is caused by either a recessive or dominant gene. Many times people are not aware that they carry the ataxia gene until they have children who begin to show signs of having the disorder.
There have been up to 60 different types of SCA identified (most are found on autopsy) as there is no one test that can tell if an individual has SCA or what type it is. Many are misdiagnosed or go years without knowing the exact type. In 2008 there was a ataxia genetic blood test developed to test for 12 of these many types. This test for the most common hereditary types of Ataxia which include, Friedreich’s ataxia, SCA 1,3,8 and a few more. However in the SCA group, with so many different types, most go with a diagnosis of SCA unidentified or unknown. Usually the diagnosis comes after examination by a neurologist, which includes a physical exam, family history, MRI scanning of the brain and spine, and spinal tap.
The following is a list of some, not all, types of Spinocerebellar ataxia. The first ataxia gene was identified in 1993 for a dominantly inherited type. It was called 鉄pinocerebellar ataxia type 1″ (SCA1). Subsequently, as additional dominant genes were found they were called SCA2, SCA3, etc. Usually, the “type” number of “SCA” refers to the order in which the gene was found. At this time, there are at least 29 different gene mutations which have been found (not all listed).
Many SCAs below fall under the category of polyglutamine diseases, which are caused when a disease-associated protein (i.e. ataxin-1, ataxin-3, etc.) contains a glutamine repeat beyond a certain threshold. In most dominant polyglutamine diseases, the glutamine repeat threshold is approximately 35, except for SCA3 which is beyond 50. Polyglutamine diseases are also known as “CAG Triplet Repeat Disorders” because CAG is the codon which codes for the amino acid glutamine. Many prefer to refer to these also as polyQ diseases since “Q” is the one-letter reference for glutamine.
Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements. This frequent hand movements cause intentional tremor in these patients. Frequently, atrophy of the cerebellum occurs, and different ataxias are known to affect different regions within the cerebellum. As with other forms of ataxia, SCA results in unsteady and clumsy motion of the body due to a failure of the fine coordination of muscle movements, along with other symptoms.
The symptoms of an ataxia vary with the specific type and with the individual patient. Generally, a person with ataxia retains full mental capacity but may progressively lose physical control.
The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. The hereditary ataxias can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
Many types of autosomal dominant cerebellar ataxias are now known for which specific genetic information is available. Synonyms for autosomal dominant cerebellar ataxias (ADCA) used prior to the current understanding of the molecular genetics were Marie’s ataxia, inherited olivopontocerebellar atrophy, cerebello-olivary atrophy, or the more generic term “spinocerebellar degeneration.” (Spinocerebellar degeneration is a rare inherited neurological disorder of the central nervous system characterized by the slow degeneration of certain areas of the brain. There are three forms of spinocerebellar degeneration: Types 1, 2, 3. Symptoms begin during adulthood.)
There are five typical autosomal recessive disorders in which ataxia is a prominent feature: Friedreich ataxia, ataxia-telangiectasia, ataxia with vitamin E deficiency, ataxia with oculomotor apraxia (AOA), spastic ataxia. Disorder subdivisions: Friedreich’s ataxia, Spinocerebellar ataxia, Ataxia telangiectasia, Vasomotor ataxia, Vestibulocerebellar, Ataxiadynamia, Ataxiophemia, Olivopontocerebellar atrophy, and Charcot-Marie-Tooth disease.
There have been reported cases where a polyglutamine expansion may lengthen when passed down, which often can result in an earlier age-of-onset and a more severe disease phenotype for individuals who inherit the disease allele. This falls under the category of genetic anticipation.
There is no currently known cure for spinocerebellar ataxia, which is considered to be a progressive and irreversible disease, although not all types cause equally severe disability. Treatments are generally directed towards alleviating symptoms, not the disease itself. Many patients with hereditary or idiopathic forms of ataxia have other symptoms in addition to ataxia. Medications or other therapies might be appropriate for some of these symptoms, which could include tremor, stiffness, depression, spasticity, and sleep disorders, among others. Both onset of initial symptoms and duration of disease are variable. If the disease is caused by a polyglutamine trinucleotide repeat CAG expansion, a longer expansion may lead to an earlier onset and a more radical progression of clinical symptoms. Typically, a person afflicted with this disease will eventually be unable to perform daily tasks (ADLs). However, rehabilitation therapists can help patients to maximize their ability of self-care and delay deterioration to certain extent. Stem cell research has been sought for a future treatment.
Physical therapists can assist patients in maintaining their level of independence through therapeutic exercise programs. Physical therapy generally emphasizes postural balance and gait training for ataxia patients. General conditioning such as range-of-motion exercises and muscle strengthening would also be included in therapeutic exercise programs. Research showed that spinocerebellar ataxia 2 (SCA2) patients with a mild stage of the disease gained significant improvement in static balance and neurological indices after six months of a physical therapy exercise training program. Occupational therapists may assist patients with incoordination or ataxia issues through the use of adaptive devices. Such devices may include a cane, crutches, walker, or wheelchair for those with impaired gait. Other devices are available to assist with writing, feeding, and self care if hand and arm coordination are impaired. A randomized clinical trial revealed that an intensive rehabilitation program with physical and occupational therapies for patients with degenerative cerebellar diseases can significantly improve functional gains in ataxia, gait and activities of daily living. Some level of improvement was shown to be maintained 24 weeks post-treatment. Speech language pathologists may use augmentative and alternative communication devices to help patients with impaired speech.

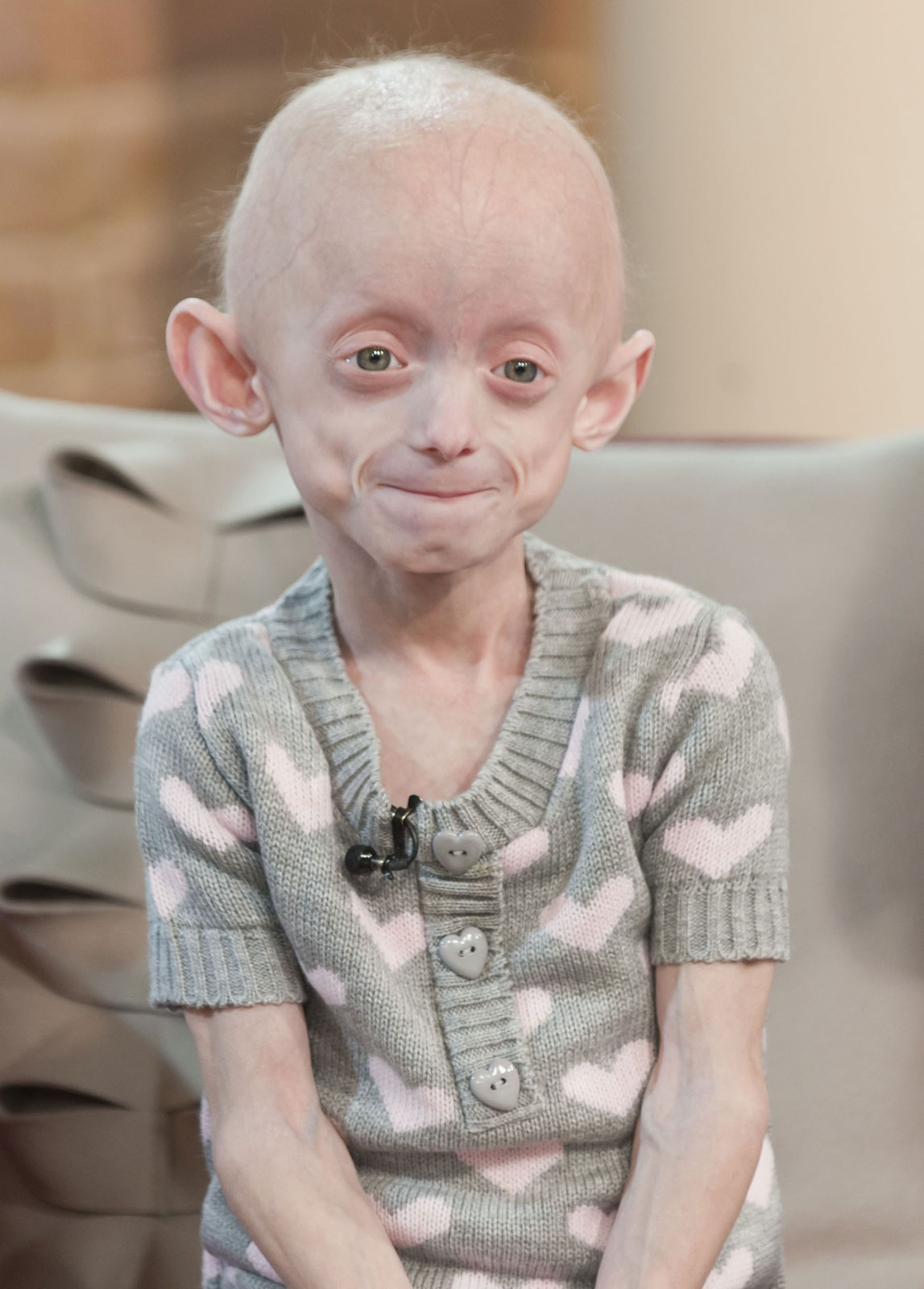
Progeria is a disease that causes children to age very prematurely and quickly. A baby with the genetic mutation is born a normal, healthy baby but symptoms normally appear when the child is 18-24 months. Children with progeria have bodies like those of elderly people, with wrinkled skin and poor eyesight and very few sufferers live beyond 13 years.

Progeria (Hutchinson-Gilford Progeria Syndrome, HGPS, Progeria syndrome) is an extremely rare genetic disease wherein symptoms resembling aspects of aging are manifested at a very early age. Progeria is one of several progeroid syndromes. The word progeria comes from the Greek words “pro” (p??), meaning “before” or “premature”, and “geras” (???a?), meaning “old age”. The disorder has a very low incident rate, occurring in an estimated 1 per 8 million live births. Those born with progeria typically live to their mid teens to early twenties. It is a genetic condition that occurs as a new mutation, and is rarely inherited. Although the term progeria applies strictly speaking to all diseases characterized by premature aging symptoms, and is often used as such, it is often applied specifically in reference to Hutchinson-Gilford Progeria Syndrome (HGPS).
Scientists are particularly interested in progeria because it might reveal clues about the normal process of aging. Progeria was first described in 1886 by Jonathan Hutchinson. It was also described independently in 1897 by Hastings Gilford. The condition was later named Hutchinson-Gilford Progeria Syndrome.
Children with progeria usually develop the first symptoms during their first few months. The earliest symptoms may include a failure to thrive and a localized scleroderma-like skin condition. As a child ages past infancy, additional conditions become apparent usually around 18–24 months. Limited growth, full-body alopecia, and a distinctive appearance (a small face with a shallow recessed jaw, and a pinched nose) are all characteristics of progeria. Signs and symptoms of this progressive disease tend to get worse as the child ages. Later, the condition causes wrinkled skin, atherosclerosis, kidney failure, loss of eyesight, hair loss, and cardiovascular problems. Scleroderma, a hardening and tightening of the skin on trunk and extremities of the body, is prevalent. People diagnosed with this disorder usually have small, fragile bodies, like those of elderly people. The face is usually wrinkled, with a larger head in relation to the body, a narrow face and a beak nose. Prominent scalp veins are noticeable (made more obvious by alopecia), as well as prominent eyes. Musculoskeletal degeneration causes loss of body fat and muscle, stiff joints, hip dislocations, and other symptoms generally absent in the non-elderly population. Individuals do usually retain normal mental and motor development.
In normal conditions, the LMNA gene codes for a structural protein called prelamin A. There is a farnesyl functional group attached to the carboxyl-terminus of its structure. The farnesyl group allows prelamin A to attach temporarily to the nuclear rim. Once the protein is attached, the farnesyl group is removed. Failure to remove this farnesyl group, permanently affixes the protein to the nuclear rim. Without its farnesyl group, prelamin A is referred to as lamin A. Lamin A, along with lamin B and lamin C, make up the nuclear lamina, which provides structural support to the nucleus.
Before the late 20th century, research on progeria yielded very little information about the syndrome. In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, in which cytosine is replaced with thymine. This mutation causes transcription of the LMNA gene to stop too early, which results in the creation of an abnormally short mRNA transcript. This mRNA strand, when translated, yields an abnormal variant of the prelamin A protein whose farnesyl group cannot be removed. Because its farnesyl group cannot be removed, this abnormal protein, referred to as progerin, is permanently affixed to the nuclear rim, and therefore does not become part of the nuclear lamina. Without lamin A, the nuclear lamina is unable to provide the nuclear envelope with adequate structural support, causing it to take on an abnormal shape. Since the support that the nuclear lamina normally provides is necessary for the organizing of chromatin during mitosis, weakening of the nuclear lamina limits the ability of the cell to divide.
Progerin may also play a role in normal human aging, since its production is activated in senescent wildtype cells. Unlike “accelerated aging diseases” (such as Werner’s syndrome, Cockayne’s syndrome, or xeroderma pigmentosum), progeria is not caused by defective DNA repair. Because these diseases cause changes in different aspects of aging, but never in every aspect, they are often called “segmental progerias”.
Diagnosis is suspected according to signs and symptoms, such as skin changes, abnormal growth, and loss of hair. A genetic test for LMNA mutations can confirm the diagnosis of progeria.
No treatments have been proven effective. Most treatment focuses on reducing complications (such as cardiovascular disease) with heart bypass surgery or low-dose aspirin. Children may also benefit from a high-energy diet. Growth hormone treatment has been attempted. The use of morpholinos has also been attempted in order to reduce progerin production. Antisense Morpholino oligonucleotides specifically directed against the mutated exon 11–exon 12 junction in the mutated pre-mRNAs were used.
A type of anticancer drug, the farnesyltransferase inhibitors (FTIs), has been proposed, but their use has been mostly limited to animal models. A Phase II clinical trial using the FTI lonafarnib began in May 2007. In studies on the cells another anti-cancer drug, rapamycin, caused removal of progerin from the nuclear membrane through autophagy. It has been proved that pravastatin and zoledronate are effective drugs when it comes to the blocking of farnesyl group production. However, it is important to remember that no treatment is able to cure progeria.
Farnesyltransferase inhibitors (FTIs) are drugs which inhibit the activity of an enzyme needed in order to make a link between progerin proteins and farnesyl groups. This link generates the permanent attachment of the progerin to the nuclear rim. In progeria, cellular damage can be appreciated because that attachment takes place and the nucleus is not in a normal state. Lonafarnib is an FTI, which means it can avoid this link, so progerin can not remain attached to the nucleus rim and it now has a more normal state. The delivery of Lonafarnib is not approved by the US Food and Drug Administration (FDA). Therefore, it can only be used in certain clinical trials. Until the treatment of FTIs is implemented in progeria children we will not know its effects—which are positive in mice.

Pravastatin, traded as Pravachol or Selektine, is included in the family of statins. As well as zoledronate (also known as Zometa and Reclast, which is a bisphosphonate), its utility in HGPS is the prevention of farnesyl groups formation, which progerin needs to provoke the disease. Some animal trials have been realized using FTIs or a combination of pravastatin and zoledronate so as to observe whether they are capable of reversing abnormal nuclei. The results, obtained by blinded electron microscopic analysis and immunofluorescence microscopy, showed that nucleus abnormalities could be reversed in transgenic mice expressing progerin. The reversion was also observed in vivo—cultured cells from human subjects with progeria—due to the action of the pharmacs, which block protein prenylation (transfer of a farnesyl polypeptide to C-terminal cysteine).

The authors of that trial add, when it comes to the results, that: “They further suggest that skin biopsy may be useful to determine if protein farnesylation inhibitors are exerting effects in subjects with HGPS in clinical trials”. Unlike FTIs, pravastatin and zoledronate were approved by the U.S. FDA (in 2006 and 2001 respectively), although they are not sold as a treatment for progeria. Pravastatin is used to decrease cholesterol levels and zoledronate to prevent hypercalcaemia.
Rapamycin, also known as Sirolimus, is a macrolide. There are recent studies concerning rapamycin which conclude that it can minimize the phenotypic effects of progeria fibroblasts. Other observed consequences of its use are: abolishment of nuclear blebbing, degradation of progerin in affected cells and reduction of insoluble progerin aggregates formation. All these results do not come from any clinical trial, although it is believed that the treatment might benefit HGPS kids. A 2012 study showed that the cancer drug Lonafarnib can be used to treat progeria. It should always be taken in account that no treatment is delivered in order to cure Hutchinson-Gilford progeria syndrome; all potential drugs are in pre-clinical stages.
Several discoveries have been made that have led to greater understandings and perhaps eventual treatment for this disease. A 2003 report in Nature said that progeria may be a de novo dominant trait. It develops during cell division in a newly conceived zygote or in the gametes of one of the parents. It is caused by mutations in the LMNA (lamin A protein) gene on chromosome 1; the mutated form of lamin A is commonly known as progerin. One of the authors, Leslie Gordon, was a physician who did not know anything about progeria until her own son, Sam, was diagnosed at 21 months. Gordon and her husband, pediatrician Scott Berns, founded the Progeria Research Foundation.

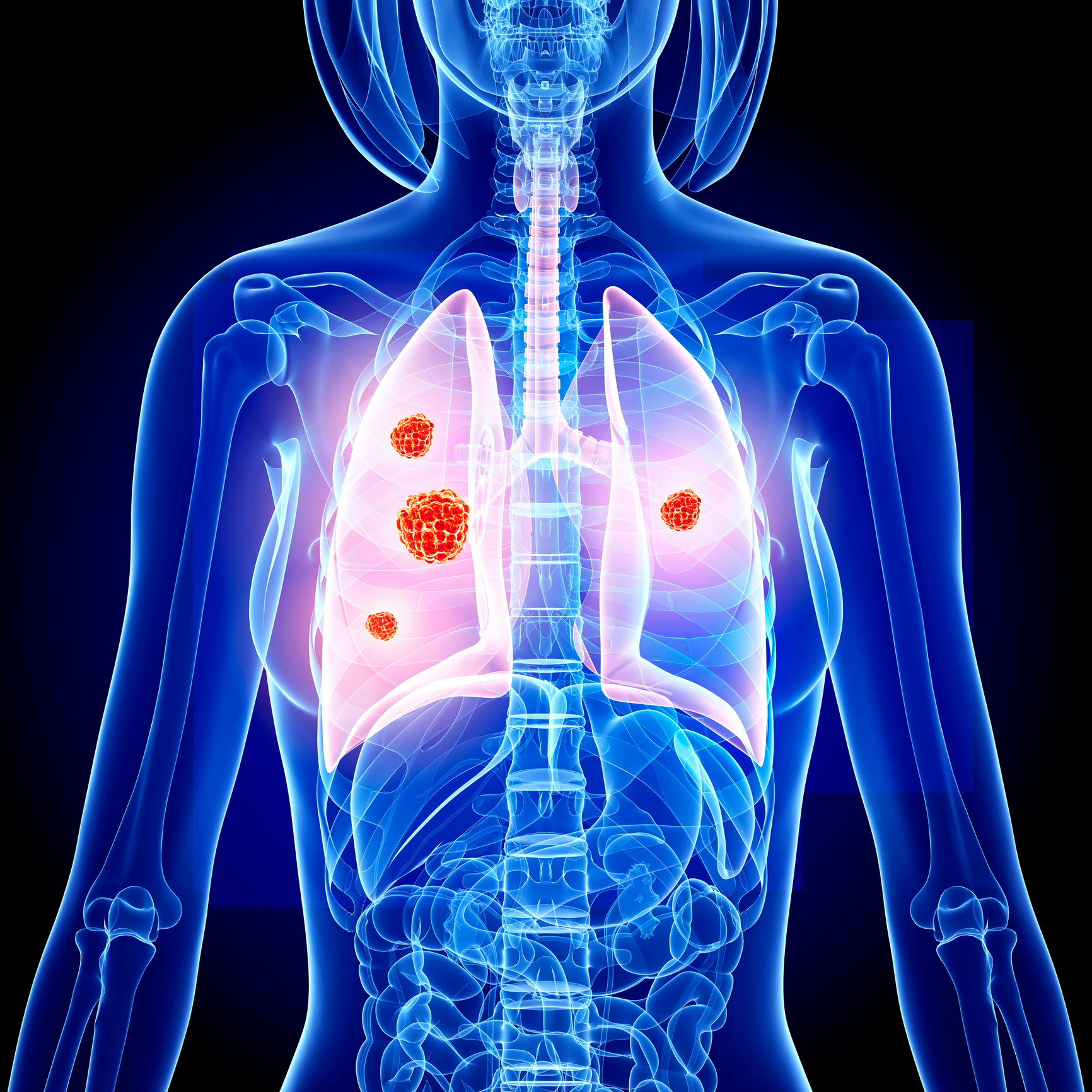
Lung cancer is a deadly disease in which the average survival is 2 to 4 months without treatment, says MedlinePlus. It states that 15 percent of lung cancers are of the small cell type. This type of lung cancer can rapidly spread and create havoc in the body.

Symptoms of small cell lung cancer include shortness of breath, wheezing, coughing up blood, unintentional weight loss and chest pain. This type of lung cancer can also lead to a poor appetite, fever, hoarseness, difficulty swallowing and facial swelling. In some cases, it can lead to weakness and changes in the voice. Small cell lung cancer typically occurs as a result of smoking.
Chemotherapy medications such as etopside, along with radiation treatment may help manage the symptoms of small cell lung cancer but does not cure it. MedlinePlus says that surgery only helps a few people as small cell lung cancer typically spreads by the time it is discovered.
Small-cell carcinoma (sometimes known as “small-cell lung cancer”, or “oat-cell carcinoma”) is a type of highly malignant cancer that most commonly arises within the lung, although it can occasionally arise in other body sites, such as the cervix, prostate, and gastrointesinal tract.
Small-cell carcinoma is an undifferentiated neoplasm composed of primitive-appearing cells.
As the name implies, the cells in small-cell carcinomas are smaller than normal cells, and barely have room for any cytoplasm. Some researchers identify this as a failure in the mechanism that controls the size of the cells.
In a significant number of cases, small-cell carcinomas can produce ectopic hormones, including adrenocorticotropic hormone (ACTH) and anti-diuretic hormone (ADH). Ectopic production of large amounts of ADH leads to syndrome of inappropriate antidiuretic hormone hypersecretion (SIADH). Lambert-Eaton myasthenic syndrome (LEMS) is a well-known paraneoplastic condition linked to small-cell carcinoma.
Histopathologic image of small-cell carcinoma of the lung. CT-guided core needle biopsy. H&E stain. When associated with the lung, it is sometimes called “oat cell carcinoma” due to the flat cell shape and scanty cytoplasm. It is thought to originate from neuroendocrine cells (APUD cells) in the bronchus called Feyrter cells (named for Friedrich Feyrter). Hence, they express a variety of neuroendocrine markers, and may lead to ectopic production of hormones like ADH and ACTH that may result in paraneoplastic syndromes and Cushing’s syndrome. Approximately half of all individuals diagnosed with Lambert-Eaton myasthenic syndrome (LEMS) will eventually be found to have a small-cell carcinoma of the lung.
Small-cell carcinoma is most often more rapidly and widely metastatic than non-small cell lung carcinoma (and hence staged differently). There is usually early involvement of the hilar and mediastinal lymph nodes.
Combined small-cell lung carcinoma (c-SCLC)
Small-cell lung carcinoma can occur in combination with a wide variety of other histological variants of lung cancer, including extremely complex malignant tissue admixtures. When it is found with one or more differentiated forms of lung cancer, such as squamous cell carcinoma or adenocarcinoma, the malignant tumor is then diagnosed and classified as a combined small cell lung carcinoma (c-SCLC). C-SCLC is the only currently recognized subtype of SCLC. Although combined small-cell lung carcinoma is currently staged and treated similarly to “pure” small-cell carcinoma of the lung, recent research suggests surgery might improve outcomes in very early stages of this tumor type.
Smoking is a significant etiological factor.
Symptoms and signs are as for other lung cancers. In addition, because of their neuroendocrine cell origin, small-cell carcinomas will often secrete substances that result in paraneoplastic syndromes such as Lambert-Eaton myasthenic syndrome.
Extrapulmonary small-cell carcinoma
Very rarely, the primary site for small-cell carcinoma is outside of the lungs and pleural space, it is referred to as extrapulmonary small-cell carcinoma (EPSCC). Outside of the respiratory tract, small cell carcinoma can appear in the cervix, prostate, liver, pancreas, gastrointestinal tract, or bladder. It is estimated to account for 1,000 new cases a year in the U.S. Histologically similar to small-cell lung cancer, therapies for small-cell lung cancer are usually used to treat EPSCC. First line treatment is usually with cisplatin and etoposide. In Japan, the first line treatment is shifting to irinotecan and cisplatin. When the primary site is in the skin, it is referred to as Merkel cell carcinoma.
Small-cell carcinoma of the prostate

In the prostate, small-cell carcinoma (SCCP) is a rare form of cancer (approx 1% of PC). Due to the fact that there is little variation in prostate specific antigen levels, this form of cancer is normally diagnosed at an advanced stage, after metastasis. It can metastasize to the brain.
Small-cell lung carcinoma has long been divided into two clinicopathological stages, including limited stage (LS) and extensive stage (ES). The stage is generally determined by the presence or absence of metastases, whether or not the tumor appears limited to the thorax, and whether or not the entire tumor burden within the chest can feasibly be encompassed within a single radiotherapy portal. In general, if the tumor is confined to one lung and the lymph nodes close to that lung, the cancer is said to be LS. If the cancer has spread beyond that, it is said to be ES.
In cases of LS-SCLC, combination chemotherapy (often including cyclophosphamide, cisplatinum, doxorubicin, etoposide, vincristine and/or paclitaxel) is administered together with concurrent chest radiotherapy (RT). Chest RT has been shown to improve survival in LS-SCLC.
Exceptionally high objective initial response rates (RR) of between 60% and 90% are seen in LS-SCLC using chemotherapy alone, with between 45% and 75% of individuals showing a “complete response” (CR), which is defined as the disappearance of all radiological and clinical signs of tumor. Unfortunately, relapse is the rule, and median survival is only 18 to 24 months.
Because SCLC usually metastasizes widely very early on in the natural history of the tumor, and because nearly all cases respond dramatically to CT and/or RT, there has been little role for surgery in this disease since the 1970s. However, recent work suggests that in cases of small, asymptomatic, node-negative SCLC’s (“very limited stage”), surgical excision may improve survival when used prior to chemotherapy.(“adjuvant chemotherapy”).
In ES-SCLC, combination chemotherapy is the standard of care, with radiotherapy added only to palliate symptoms such as dyspnea, pain from liver or bone metastases, or for treatment of brain metastases, which, in small-cell lung carcinoma, typically have a rapid, if temporary, response to whole brain radiotherapy.
Combination chemotherapy consists of a wide variety of agents, including cisplatin, cyclophosphamide, vincristine and carboplatin. Response rates are high even in extensive disease, with between 15% and 30% of subjects having a complete response to combination chemotherapy, and the vast majority having at least some objective response. Responses in ES-SCLC are often of short duration, however.
If complete response to chemotherapy occurs in a subject with SCLC, then prophylactic cranial irradiation (PCI) is often used in an attempt to prevent the emergence of brain metastases. Although this treatment is often effective, it can cause hair loss and fatigue. Prospective randomized trials with almost two years follow-up have not shown neurocognitive ill-effects. Meta-analyses of randomized trials confirm that PCI provides significant survival benefits. All in all, small-cell carcinoma is very responsive to chemotherapy and radiotherapy, and in particular, regimens based on platinum-containing agents. However, most people with the disease relapse, and median survival remains low.
In limited-stage disease, median survival with treatment is 14–20 months, and about 20% of patients with limited-stage small-cell lung carcinoma live 5 years or longer. Because of its predisposition for early metastasis, the prognosis of SCLC is poor with only 10% to 15% of patients surviving 3 years (Washington and Leaver, 2010).
The prognosis is far worse in extensive-stage small-cell lung carcinoma, with treatment, median survival is just 8–13 months, and only 1–5% of patients with extensive-stage small-cell lung carcinoma treated with chemotherapy live 5 years or longer.


Also often referred to simply as lupus, this is an autoimmune disorder that causes chronic inflammation in various parts of the body. Three main types of lupus are recognized—discoid, systemic, and drug-induced.Discoid lupus affects only the skin and does not usually involve internal organs. The term discoid refers to a rash of distinct reddened patches covered with grayish brown scales that may appear on the face, neck, and scalp. In about 10 percent of people with discoid lupus, the disease will evolve into the more severe systemic form of the disorder.Systemic lupus erythematosus is the most common form of the disease. It may affect virtually any organ or structure of the body, especially the skin, kidneys, joints, heart, gastrointestinal tract, brain, and serous membranes (membranous linings of organs, joints, and cavities of the body.) While systemic lupus can affect any area of the body, most people experience symptoms in only a few organs. The skin rash, if present, resembles that of discoid lupus. In general, no two people will have identical symptoms. The course of the disease is also variable and is marked by periods when the disease is active and by other periods when symptoms are not evident (remission).
Lupus erythematosus is a name given to a collection of autoimmune diseases, in which the human immune system becomes hyperactive and attacks normal, healthy tissues. Symptoms of these diseases can affect many different body systems, including joints, skin, kidneys, blood cells, heart, and lungs.
Symptoms vary from person to person, and may come and go. Almost everyone with Lupus has joint pain and swelling. Some develop arthritis. Frequently affected joints are the fingers, hands, wrists, and knees. Other common symptoms include:
- Chest pain when taking a deep breath
- Fatigue
- Fever with no other cause
- General discomfort, uneasiness, or ill feeling (malaise)
- Hair loss
- Mouth sores
- Sensitivity to sunlight
- Skin rash – a “butterfly” rash in about half people with SLE.
- Swollen lymph nodes

Photosensitivity is a known symptom of lupus, but its relationship to and influence on other aspects of the disease remain to be defined. Causes of photosensitivity may include:
change in autoantibody location

cytotoxicity
inducing apoptosis with autoantigens in apoptotic blebs
upregulation of adhesion molecules and cytokines
inducing nitric oxide synthase expression
ultraviolet-generated antigenic DNA.
Tumor necrosis factor alpha also seems to play a role in the development of photosensitivity.
Treatment consists primarily of immunosuppressive drugs (e.g., hydroxychloroquine and corticosteroids). In 2011, the U.S. Food and Drug Administration (FDA) approved the first new drug for Lupus in more than 50 years to be used in the US, Belimumab
An estimated 5 million people worldwide have some form of Lupus.
70% of Lupus cases diagnosed are Systemic Lupus Erythematosus.
20%of people with lupus will have a parent or sibling who already has lupus or may develop lupus.
About 5% of the children born to individuals with lupus will develop the illness.
United Kingdom
Systemic Lupus Erythematosus
Affects UK females far more than males at a ratio of 7:1, that is to say females are 7 times more likely to have the disease.
The estimated number of UK females with SLE is 21,900, and UK males with Lupus is 3000 – a total of 24,700, or 0.041% of the population.
SLE is more common amongst certain ethnic groups than others, especially those of African origin.
United States
Systemic Lupus Erythematosus:
Occurs from infancy to old age, with peak occurrence between ages 15 and 40.
Affects US females far more than males at a ratio of 6-10:1, that is to say females are 6-10 times more likely to have the disease.
Prevalence data is limited. Estimates vary and range from 1.8 to 7.6 cases per 100,000 persons per year in parts of the continental United States.
Lupus is two to three times more prevalent among women of color – African Americans, Hispanics/Latinos, Asians, Native Americans, Alaska Natives, Native Hawaiians and other Pacific Islanders – than among Caucasian women.