 Spinocerebellar ataxia
Spinocerebellar ataxia
 This degenerative incurable disease is genetic and there are many different types of it. While there is no cure for it, there are drugs that can help to alleviate the symptoms, which are typically loss of muscle control and tremors. Some sufferers can have a normal lifespan while others can die very young.
This degenerative incurable disease is genetic and there are many different types of it. While there is no cure for it, there are drugs that can help to alleviate the symptoms, which are typically loss of muscle control and tremors. Some sufferers can have a normal lifespan while others can die very young.
Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a disease in its own right. An estimated 150,000 people in the United States are diagnosed with Ataxia, SCA’s are the largest group of this hereditary, progressive, degenerative and often fatal neurodegenerative disorders. There is no known effective treatment or cure. Ataxia can affect anyone of any age. It is caused by either a recessive or dominant gene. Many times people are not aware that they carry the ataxia gene until they have children who begin to show signs of having the disorder.
There have been up to 60 different types of SCA identified (most are found on autopsy) as there is no one test that can tell if an individual has SCA or what type it is. Many are misdiagnosed or go years without knowing the exact type. In 2008 there was a ataxia genetic blood test developed to test for 12 of these many types. This test for the most common hereditary types of Ataxia which include, Friedreich’s ataxia, SCA 1,3,8 and a few more. However in the SCA group, with so many different types, most go with a diagnosis of SCA unidentified or unknown. Usually the diagnosis comes after examination by a neurologist, which includes a physical exam, family history, MRI scanning of the brain and spine, and spinal tap.
The following is a list of some, not all, types of Spinocerebellar ataxia. The first ataxia gene was identified in 1993 for a dominantly inherited type. It was called 鉄pinocerebellar ataxia type 1″ (SCA1). Subsequently, as additional dominant genes were found they were called SCA2, SCA3, etc. Usually, the “type” number of “SCA” refers to the order in which the gene was found. At this time, there are at least 29 different gene mutations which have been found (not all listed).
Many SCAs below fall under the category of polyglutamine diseases, which are caused when a disease-associated protein (i.e. ataxin-1, ataxin-3, etc.) contains a glutamine repeat beyond a certain threshold. In most dominant polyglutamine diseases, the glutamine repeat threshold is approximately 35, except for SCA3 which is beyond 50. Polyglutamine diseases are also known as “CAG Triplet Repeat Disorders” because CAG is the codon which codes for the amino acid glutamine. Many prefer to refer to these also as polyQ diseases since “Q” is the one-letter reference for glutamine.
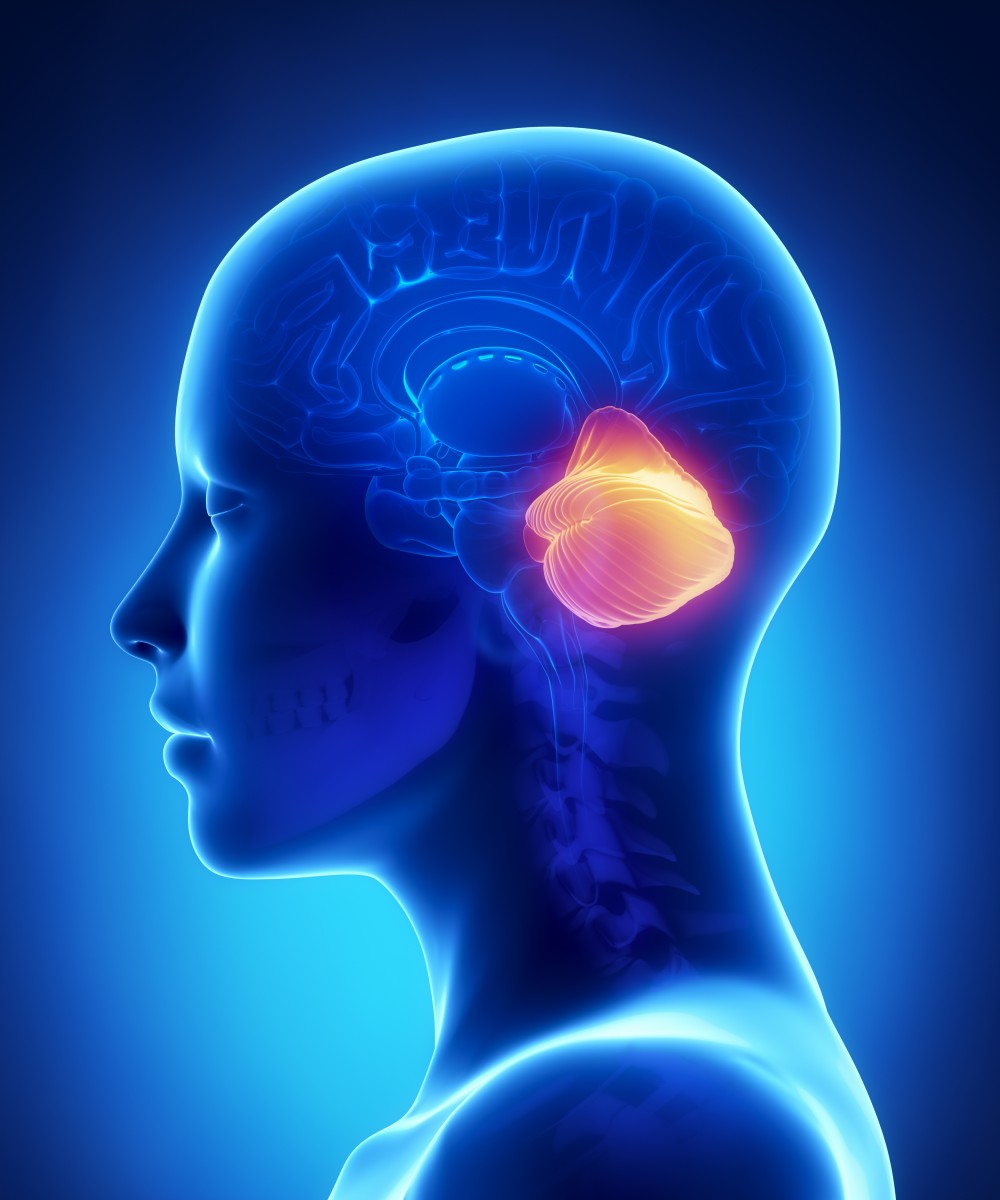
Spinocerebellar ataxia (SCA) is one of a group of genetic disorders characterized by slowly progressive incoordination of gait and is often associated with poor coordination of hands, speech, and eye movements. This frequent hand movements cause intentional tremor in these patients. Frequently, atrophy of the cerebellum occurs, and different ataxias are known to affect different regions within the cerebellum. As with other forms of ataxia, SCA results in unsteady and clumsy motion of the body due to a failure of the fine coordination of muscle movements, along with other symptoms.
The symptoms of an ataxia vary with the specific type and with the individual patient. Generally, a person with ataxia retains full mental capacity but may progressively lose physical control.
The hereditary ataxias are categorized by mode of inheritance and causative gene or chromosomal locus. The hereditary ataxias can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner.
Many types of autosomal dominant cerebellar ataxias are now known for which specific genetic information is available. Synonyms for autosomal dominant cerebellar ataxias (ADCA) used prior to the current understanding of the molecular genetics were Marie’s ataxia, inherited olivopontocerebellar atrophy, cerebello-olivary atrophy, or the more generic term “spinocerebellar degeneration.” (Spinocerebellar degeneration is a rare inherited neurological disorder of the central nervous system characterized by the slow degeneration of certain areas of the brain. There are three forms of spinocerebellar degeneration: Types 1, 2, 3. Symptoms begin during adulthood.)
There are five typical autosomal recessive disorders in which ataxia is a prominent feature: Friedreich ataxia, ataxia-telangiectasia, ataxia with vitamin E deficiency, ataxia with oculomotor apraxia (AOA), spastic ataxia. Disorder subdivisions: Friedreich’s ataxia, Spinocerebellar ataxia, Ataxia telangiectasia, Vasomotor ataxia, Vestibulocerebellar, Ataxiadynamia, Ataxiophemia, Olivopontocerebellar atrophy, and Charcot-Marie-Tooth disease.
There have been reported cases where a polyglutamine expansion may lengthen when passed down, which often can result in an earlier age-of-onset and a more severe disease phenotype for individuals who inherit the disease allele. This falls under the category of genetic anticipation.
There is no currently known cure for spinocerebellar ataxia, which is considered to be a progressive and irreversible disease, although not all types cause equally severe disability. Treatments are generally directed towards alleviating symptoms, not the disease itself. Many patients with hereditary or idiopathic forms of ataxia have other symptoms in addition to ataxia. Medications or other therapies might be appropriate for some of these symptoms, which could include tremor, stiffness, depression, spasticity, and sleep disorders, among others. Both onset of initial symptoms and duration of disease are variable. If the disease is caused by a polyglutamine trinucleotide repeat CAG expansion, a longer expansion may lead to an earlier onset and a more radical progression of clinical symptoms. Typically, a person afflicted with this disease will eventually be unable to perform daily tasks (ADLs). However, rehabilitation therapists can help patients to maximize their ability of self-care and delay deterioration to certain extent. Stem cell research has been sought for a future treatment.
Physical therapists can assist patients in maintaining their level of independence through therapeutic exercise programs. Physical therapy generally emphasizes postural balance and gait training for ataxia patients. General conditioning such as range-of-motion exercises and muscle strengthening would also be included in therapeutic exercise programs. Research showed that spinocerebellar ataxia 2 (SCA2) patients with a mild stage of the disease gained significant improvement in static balance and neurological indices after six months of a physical therapy exercise training program. Occupational therapists may assist patients with incoordination or ataxia issues through the use of adaptive devices. Such devices may include a cane, crutches, walker, or wheelchair for those with impaired gait. Other devices are available to assist with writing, feeding, and self care if hand and arm coordination are impaired. A randomized clinical trial revealed that an intensive rehabilitation program with physical and occupational therapies for patients with degenerative cerebellar diseases can significantly improve functional gains in ataxia, gait and activities of daily living. Some level of improvement was shown to be maintained 24 weeks post-treatment. Speech language pathologists may use augmentative and alternative communication devices to help patients with impaired speech.
Disclaimer
The Content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition.